| SRA Toolkit安装及使用教程 | 您所在的位置:网站首页 › selection tool安装教程 › SRA Toolkit安装及使用教程 |
SRA Toolkit安装及使用教程
|
您是否对基因组学研究充满了好奇和渴望?想要深入了解生物的遗传信息,揭示其中隐藏的奥秘?那么,SRA Toolkit将成为您最强大的助手。作为一款功能强大且易于使用的工具集,SRA Toolkit将为您提供快速、高效地获取和分析原始测序数据的解决方案。 Sratools是NCBI官方提供,用于操作SRA (reads and reference alignments) 数据的工具集合,一般常用于: 下载SRA文件,从SRA文件中提取fastq文件。SRA Toolkit的安装 1、conda 安装(推荐使用) conda install sra-tools -y2、传统安装 2.1 在Linux系统(以CentOS为例)下将上述的链接下载到本地 wget --output-document sratoolkit.tar.gz https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz2.2 解压安装包 tar -vxzf sratoolkit.tar.gz2.3 将sratoolkit路径添加到环境变量 export PATH=$PATH:$PWD/sratoolkit.3.0.0-mac64/bin2.4 验证 shell 是否能找到二进制文件 which fastq-dump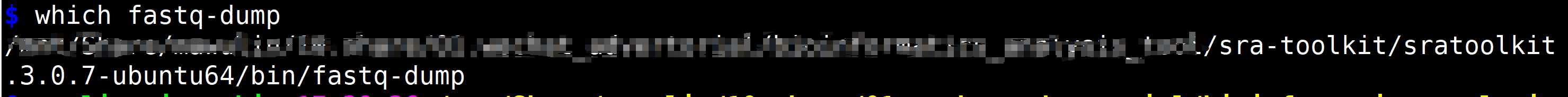 2.5 测试工具包是否正常运行(若输出以下内容则表明安装成功) fastq-dump --stdout -X 2 SRR390728 SRA Toolkit的使用 官方文档:https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc 下载SRA下载单个文件: prefetch SRR390728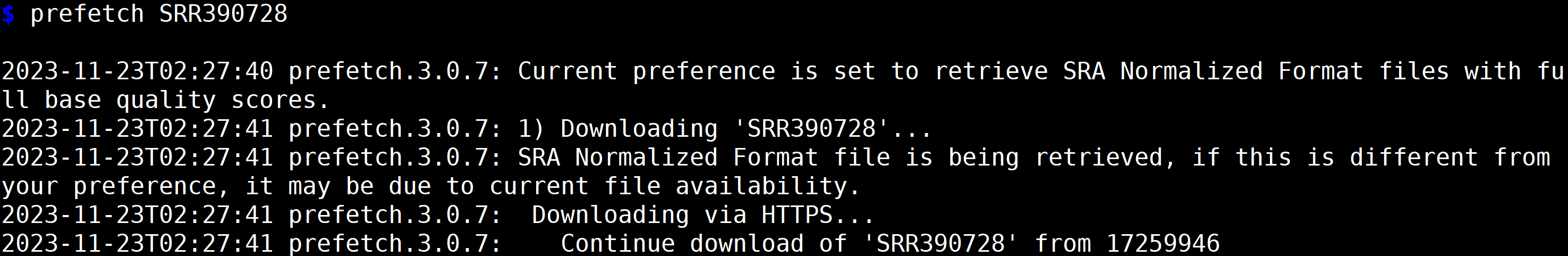 下载多个文件: # 创建一个accession文件,用于保存序列号 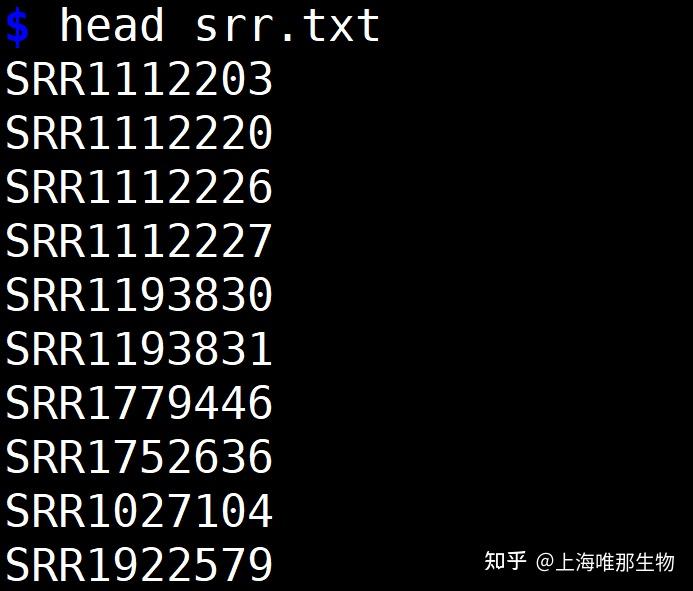 prefetch --option-file srr.txt prefetch --option-file srr.txt 2. 抽取fastq文件(在下载到的sra文件所在目录下) 2.1 fastq-dump使用 # 查看帮助文档 fastq-dump -h # 抽提fq文件,并将其压缩 fastq-dump --gzip -O $PWD --split-3 *.sra# --gzip :输出gz格式压缩文件,节省空间,稍微多费点时间 # -O ${directory} :设置输出的文件间路径,outdirectory改为相应路径 # --split-3 :不知道sra是单端还是双端,默认使用--split-3   2.2 fasterq-dump使用 # 多线程抽提fq文件 fasterq-dump -p -e 24 --split-3 -O ${outdirectory} SRR1234567.sra#-p 可以显示进程 #-e 24 使用24个线程 # fasterq-dump不能使用压缩命令。  参考文献: https://github.com/ncbi/sra-tools/wiki 本推文来源于上海唯那生物的“密码子实验室”公众号,微信公众号搜索“密码子实验室”即可关注,欢迎转发,如有相关课程问题,请联系唯那生物技术客服小唯(微信号:winnerbio01) |
【本文地址】